Fibrosi quística
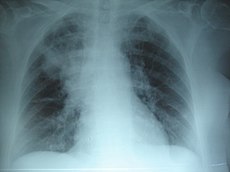 Pneumònia apical dreta: en la FQ, la infecció crònica dóna lloc a la destrucció del parènquima pulmonar, ocasionant en darrer terme la mort per insuficiència respiratòria. | |
| Tipus | malaltia autosòmica recessiva, malaltia pulmonar, genetic biliary tract disease |
|---|---|
| Especialitat | genètica mèdica |
| Medicació | fenilbutirat de sodi, Dornase alfa |
| Associació genètica | AGTR2 |
| Classificació | |
| CIM-10 | E84 |
| CIM-9 | 277 |
| CIAP | T99 |
| Recursos externs | |
| OMIM | 219700 |
| DiseasesDB | 3347 |
| MedlinePlus | 000107 |
| eMedicine | ped/535 |
| Patient UK | cystic-fibrosis-pro |
| MeSH | D003550 |
UMLS CUI | C0010674 i C0010674 |
| DOID | DOID:1485 i DOID:1485 |
La fibrosi quística (abreviatura FQ), també coneguda com a mucoviscidosi (del llatí muccus, "moc", i viscōels seus, "aferrissós"), és una malaltia hereditària freqüent que afecta a l'organisme de forma generalitzada, causant discapacitat progressiva i mort prematura. La dificultat per respirar és el símptoma més comú emergent d'infeccions pulmonars cròniques, les quals poden mostrar-se resistents al tractament amb antibiòtics i altres fàrmacs. La FQ és un trastorn multisistèmic que causa la formació i acumulació d'un moc espès i aferrissós, afectant fonamentalment pulmons, intestins, pàncrees i fetge. Així mateix, es caracteritza per la presència d'una alta concentració de sal (NaCl) en la suor, cosa que va asseure les bases de la prova estàndard per a aquest diagnòstic: l'examen d'electròlits de la suor. Aquest examen avalua, entre altres ions, els nivells de clorur excretats. Una varietat de símptomes, incloent-hi infeccions sinusals, desacceleració del creixement, diarrea i infertilitat, resulten dels efectes de la FQ sobre els diferents òrgans.
Es tracta d'una de les malalties hereditàries fatals més comunes. La seva prevalença és major entre caucàsics; una de cada 25 persones d'ascendència europea és portadora asimptomàtica d'un gen per a FQ, essent la malaltia genètica més freqüent entre aquesta població. Els afectats poden rebre diagnòstic mitjançant proves genètiques prenatals; també per perquisició nounatal o, durant la infància primerenca, per l'esmentada prova de la suor. No existeix cura per a la FQ, i la supervivència mitjana per a aquests pacients s'estima en 29 anys, arribant a valors més alts en alguns països (36,8 als EUA).[1][2] En casos severs, l'empitjorament de la malaltia pot imposar la necessitat d'un trasplantament de pulmó.
La FQ és causada per una mutació en un gen anomenat regulador de la conductància transmembrana de la fibrosi quística (CFTR, per les seves sigles en anglès). Aquest gen intervé en la producció de suor, sucs gàstrics, i moc. Encara que la majoria de les persones sanes tenen dues còpies funcionals del gen, només una és necessària per impedir el desenvolupament de fibrosi quística. La FQ es desenvolupa quan cap d'aquests gens opera normalment. En conseqüència, se la considera una malaltia autosòmica recessiva. El nom fibrosi quística es refereix als processos característics de cicatrització (fibrosi) i formació de quists dintre del pàncrees, reconeguts per primera vegada en els anys 1930.[3]
Contingut
1 Signes i símptomes
1.1 Malaltia pulmonar i sinusal
1.2 Malaltia gastrointestinal, hepàtica i pancreàtica
1.3 Malaltia endocrina i creixement
1.4 Infertilitat
2 Diagnòstic
2.1 Diagnòstic prenatal
3 Fisiopatologia
3.1 Paper de la infecció crònica en la malaltia pulmonar
3.2 Biologia molecular
4 Tractament
4.1 Antibiòtics per tractar la malaltia pulmonar
4.2 Altres mètodes per tractar la malaltia pulmonar
4.3 Tractament d'altres aspectes de la FQ
4.4 Trasplantament i teràpia gènica
5 Epidemiologia
5.1 Teories sobre la prevalença de la FQ
6 Història
7 Notes
8 Bibliografia
Signes i símptomes
La simptomatologia de la fibrosi quística varia en funció de l'edat de l'individu, el grau en què es veuen afectats òrgans específics, la terapèutica instituïda prèviament, i els tipus d'infeccions associades. Aquesta malaltia compromet a l'organisme en la seva totalitat i mostra el seu impacte sobre el creixement, la funció respiratòria, la digestió, i la reproducció. El període neonatal es caracteritza per un pobre augment de pes i per una obstrucció intestinal produïda per femta densa i voluminosa. Altres símptomes apareixen més tard, durant la infantesa i a l'inici de l'adultesa. Aquests inclouen retard del creixement, adveniment de la malaltia pulmonar, i dificultats creixents per la malabsorció de vitamines i nutrients en el tracte gastrointestinal. A més, el problema en la fertilitat es torna palpable quan s'intenta aconseguir la reproducció.
A la majoria dels nens se'ls diagnostica fibrosi quística abans del primer any de vida, quan la mucositat aferrissosa que afecta pulmons i pàncrees comença a mostrar el seu impacte. En el tracte respiratori, aquestes secrecions serveixen de brou de cultiu per a diversos bacteris responsables d'infeccions cròniques, amb deteriorament progressiu i permanent del parènquima pulmonar. Conforme s'agreuja la condició respiratòria, els pacients sofreixen hipertensió pulmonar. D'altra banda, en el pàncrees, el moc obstrueix el trànsit dels enzims sintetitzats per la glàndula i impedeix que arribin als intestins per digerir i absorbir l'aliment.
Malaltia pulmonar i sinusal

Aspergillus fumigatus, un fong comú que pot conduir a l'empitjorament de la malaltia pulmonar en persones amb FQ.
La malaltia pulmonar resulta del bloqueig de les vies aèries més petites amb el moc espès característic de la fibrosi quística. La inflamació i la infecció produeixen dany als pulmons i canvis estructurals que condueixen a una varietat de símptomes. En les etapes inicials, comunament es presenta tos incessant, producció copiosa de flegma, i una disminució en la capacitat aeròbica. Molts d'aquests símptomes ocorren quan certs bacteris (fonamentalment, Pseudomonas aeruginosa) que normalment viuen en el moc espès, creixen de forma descontrolada i causen pneumònia. En estats avançats de la FQ, els canvis en l'arquitectura del pulmó produeixen dificultats respiratòries cròniques.
Altres símptomes inclouen expectoració de sang o esput sanguinolent, dilatació crònica dels bronquis o bronquíols (bronquiectàsia), elevació de la pressió sanguínia en el pulmó, insuficiència cardíaca, sensació de no estar rebent suficient oxigen o dispnea, insuficiència respiratòria i atelèctasi; en aquest cas podria requerir-se suport ventilatori.[4] A més de les infeccions bacterianes més comunes, les persones amb FQ desenvolupen amb major facilitat altres tipus de malalties respiratòries. Entre aquestes es troba l'aspergil·losi broncopulmonar al·lèrgica, caracteritzada per una resposta d'hipersensibilitat davant un fong (floridura) ordinari del gènere Aspergillus (Aspergillus fumigatus), que aguditza els problemes respiratoris. Un altre exemple és la infecció amb el complex Mycobacterium avium (MAC), un grup de actinobactèries emparentades amb Mycobacterium tuberculosi, que pot ocasionar danys majors al pulmó, i que no respon a la terapèutica amb antibiòtics convencionals.
El moc en els sins paranasals és igualment dens i aferrissós, i també pot causar oclusió dels orificis per on els sins habitualment drenen, la qual cosa fa que s'acumulin secrecions que actuen com a brou de cultiu per als patògens abans esmentats. En aquests casos, es pot presentar dolor facial, febre, secreció nasal profusa i cefalàlgies. En les persones amb FQ, sovint s'observa un creixement sobreabundant de teixit nasal (pòlips), a conseqüència de la inflamació per infecció sinusal crònica. Aquests pòlips poden agreujar l'obstrucció de les vies respiratòries superiors i intensificar les dificultats respiratòries.[5][6]
Malaltia gastrointestinal, hepàtica i pancreàtica
Amb anterioritat a la difusió de les proves prenatal i nounatal per a fibrosis quística, era freqüent que la malaltia es detectés al constatar que el nounat no podia expulsar la seva primera femta (meconi). El meconi pot obstruir completament els intestins i causar greus trastorns. Aquesta condició, anomenada ili meconial, ocorre en el 10% dels nounats amb FQ.[7] Així mateix, és també freqüent l'associació de FQ amb protrusió de les membranes rectals internes (prolapse rectal), deguda al major volum fecal, a la malnutrició, i a l'elevació de la pressió intraabdominal per tos crònica.[8]
El moc glutinós observat en el pulmó té el seu correlat en les secrecions espesses del pàncrees, òrgan responsable de proveir sucs digestius que faciliten la descomposició química dels aliments. Aquestes secrecions impedeixen el moviment dels enzims pancreàtics cap a l'intestí i produeixen un dany irreversible en el pàncrees, sovint acompanyat d'una dolorosa inflamació (pancreatitis).[9] La deficiència d'enzims digestius es tradueix en un impediment per a absorbir els nutrients, amb la subsegüent excreció d'aquests en la femta: aquest trastorn és conegut com a malabsorció. La malabsorció condueix a la desnutrició i al retard en el creixement i desenvolupament, ambdós deguts a la baixa biodisponibilidad calòrica. Les persones amb FQ tenen, en particular, problemes per absorbir les vitamines A, D, I, i K. A més de l'afecció pancreàtica, solen experimentar acidesa crònica, xerostomia, obstrucció intestinal per intussuscepció, i restrenyiment.[10] Els pacients de més edat desenvolupen també la síndrome d'obstrucció intestinal distal causada per la femta glutinosa.[11]
Aquestes secrecions també poden causar problemes en el fetge. La bilis, produïda per aquesta víscera per facilitar la digestió, podria bloquejar les vies biliars, danyant així els teixits adjacents. Amb el temps, aquesta situació condueix a la cirrosi. En aquest cas, resulten compromeses funcions de primer ordre, tals com les implicades en la neutralització de toxines, i en la síntesi d'importants proteïnes (per exemple, els factors de coagulació, responsables de la coagulació sanguínia).[12]
Malaltia endocrina i creixement
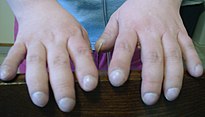
Les persones amb FQ sovint presenten malformació i eixamplament dels dits (dits en baqueta de timbal o hipocràtics)
El pàncrees conté els illots de Langerhans, que són els responsables de produir insulina, una hormona que ajuda a regular els nivells de glucosa en sang. Un dany en el pàncrees pot provocar la pèrdua de les cèl·lules dels illots i conduir a la diabetis.[13] D'altra banda, la vitamina D suplementada per l'alimentació està implicada en la regulació del calci i del fòsfor. La baixa disponibilitat de calci a causa de la malabsorció condueix a l'osteoporosi, augmentant el risc de patir fractures.[14] Addicionalment, les persones amb FQ sovint presenten, en mans i peus, una malformació denominada dits en escuradents de tambor, la qual es deu a l'efecte de la hipòxia en els ossos.
El retard en el creixement és un segell distintiu d'aquesta malaltia. Els nens amb FQ no aconsegueixen, en general, guanyar pes i altura en taxes comparables als nens de la seva edat; sovint, només reben diagnòstic apropiat una vegada que s'investiguen les causes d'aquest fenomen. Els determinants del retard en el creixement són multifactorials i inclouen la infecció pulmonar crònica, la malabsorció de nutrients en el tracte gastrointestinal, i l'augment de la demanda metabòlica associada a l'afecció crònica.
Infertilitat
| Valor normal | Fibrosi quística | |
|---|---|---|
pH | >8 | <7 |
Àcid cítric | 400-1.500 mg/100 ml | > 2.000 mg/100 ml |
Fosfatasa àcida | 140-290 µg/ml | 760-1.140 µg/ml |
Fructosa | 250–720 mg/100ml | 30–80 mg/100ml |
La infertilitat es manifesta tant entre els homes com entre les dones amb FQ. Almenys un 97% dels homes afectats són estèrils.[15] Aquests produeixen esperma normalment, però manquen del conducte deferent, que connecta els testicles amb els conductes ejaculadors del penis.[16] En molts casos d'absència congènita del conducte deferent es van trobar, durant l'examen per infertilitat, formes lleus de FQ, que no havien estat diagnosticades prèviament.[17] Un 20% de les dones amb FQ són estèrils com a conseqüència del moc cervical abundant i espès, que interfereix amb el pas de l'esperma. D'altra banda, en casos severs, la malnutrició produeix alteracions en l'ovulació i causa amenorrea.[18]
Diagnòstic

La fibrosis quística pot diagnosticar-se per tamisatge en nounats, examen d'electròlits de la suor, o prova genètica. L'any 2006, als Estats Units, el 10% dels casos van ser detectats poc després del naixement com a part dels programes de perquisició nounatal, que identifiquen nivells elevats en l'enzim tripsina. No obstant això, en la majoria dels països aquests exàmens no es realitzen de forma rutinària. Per aquesta causa, és freqüent que els afectats només rebin un diagnòstic apropiat una vegada que els símptomes forcen una avaluació per a aquesta malaltia. La prova diagnòstica més comunament utilitzada és l'examen de la suor, descrit per Lewis I. Gibson i Robert I. Cooke el 1959,[19] que usa electroforesi quantitativa (iontoforesis) amb un fàrmac estimulant de la sudoració (pilocarpina). Aquesta substància, que posseeix càrrega positiva, s'aplica sobre un elèctrode positiu (+), en contacte amb la pell. Després, mitjançant el pas de corrent elèctric, la droga migra pel tegument cap a altre elèctrode de càrrega oposada (-), col·locat a certa distància, fins a travessar l'epidermis, produint l'estimulació de les glàndules sudorípares i causant una sudoració controlada. Les mostres de suor són després col·lectades en paper de filtre o en un tub capil·lar i són analitzades, determinant-ne les concentracions de sodi i clorur. Les persones amb FQ posseeixen nivells més alts d'aquests ions en la suor. Una vegada que l'examen de la suor ha donat positiu, es realitza un diagnòstic més detallat i precís, mitjançant la identificació de les mutacions en el gen CFTR.[20]
Existeixen diverses proves per identificar eventuals complicacions i controlar l'evolució de la malaltia. Les imatges obtingudes per raigs X i TC faciliten la detecció de signes de lesió o infecció en els pulmons. El cultiu d'esput, examinat per microscopi, dona informació sobre quins són els bacteris responsables, i permet escollir els antibiòtics més efectius. Les proves de funció pulmonar medeixen les capacitats pulmonars, els volums pulmonars i la rapidesa amb què aquests poden ser mobilitzats (fluxos aeris). Per mitjà d'aquests exàmens, és possible determinar si és procedent un tractament amb antibiòtics o bé avaluar la resposta al mateix. Les anàlisis de sang poden identificar problemes hepàtics, deficiències vitamíniques, i revelar la irrupció de la diabetis. Els dispositius DEXA o DXA (sigles en l'anglès per "absorciometria de raigs X d'energia dual"), s'utilitzen com a prova per determinar la presència d'osteoporosi. Finalment, la quantificació d'elastasa fecal facilita la detecció d'insuficiència d'enzims digestius.
Diagnòstic prenatal
Les parelles que estan travessant un embaràs o que tenen plans de fer-ho, poden ser avaluades a la recerca de mutacions del gen CFTR, amb l'objectiu de determinar les probabilitats que el seu fill neixi amb fibrosi quística. La prova se sol realitzar en un dels pares o en ambdós i, en cas de detectar-se un risc elevat de FQ, s'efectua també en el fetus. Degut al fet que el diagnòstic prenatal no habilita formes de tractament superiors o alternatives, la principal raó per la qual es porta a terme és, en la pràctica, proporcionar la possibilitat d'avortament en cas que el fetus presenti la malaltia. La prova per a fibrosi quística en parelles s'ofereix de manera generalitzada en països com els Estats Units,[21] on el Col·legi Americà d'Obstetres i Ginecòlegs (ACOG, per les seves sigles en anglès) recomana la prova en parelles que posseeixen un historial de FQ entre els seus familiars directes o parents propers, i també en aquelles amb risc elevat a causa de la seva filiació ètnica.[22]
Degut al fet que el desenvolupament de la FQ en el fetus requereix que cada pare transmeti una còpia del gen CFTR mutant, i també a l'alt cost de l'examen prenatal, la prova sol realitzar-se, inicialment, només en un dels progenitors. Si aquest resulta ser portador d'una mutació del gen CFTR, llavors s'examina a l'altre per determinar el risc que el seu fill tingui la malaltia. La FQ pot resultar de més d'un miler de mutacions diferents i, amb data de 2006, no és possible efectuar estudis de laboratori per a cadascuna d'elles. La prova consisteix en analitzar la sang a la recerca de les mutacions més comunes, com la ΔF508 —la majoria de les modalitats disponibles comercialment no detecten més de 32 variants diferents. Si es coneix la dada que una família té una mutació poc comuna, aquesta pot buscar-se específicament. Com a conseqüència que no totes les mutacions conegudes són detectades per les proves corrents, un resultat negatiu no garanteix que el nadó vagi a estar lliure de la malaltia.[23] D'altra banda, atès que les mutacions sondejades són necessàriament aquelles més comunes en els grups de més alt risc, les proves en ètnies de baix risc són menys exitoses, ja que les mutacions més esteses en aquests grups són menys freqüents en la població general.
Mapa citogenètic o cariograma d'una nena, resultat d'una amniocentesi.
Les parelles en situació de risc sovint realitzen proves addicionals abans o durant l'embaràs. La fecundació in vitro amb diagnòstic genètic preimplantacional ofereix la possibilitat d'examinar l'embrió abans de la seva col·locació en l'úter. Aquesta prova es realitza tres dies després de la fecundació i procura determinar la presència de gens CFTR anormals. Si, en un embrió, resulten identificats dos gens CTRF mutants, aquest serà exclòs de la transferència, i s'implantarà un que compti amb, almenys, un gen normal.
Durant el transcurs de l'embaràs, és possible realitzar proves tant de la placenta (mostra de vellositat coriònica) com del líquid amniòtic que envolta el fetus (amniocentesi), amb l'ajut de l'ultrasò. No obstant això, la biòpsia de vellositats coriòniques es correlaciona amb el risc de mort fetal en una taxa d'1 en 100, i l'amniocentesis, d'1 en 200,[24] pel que és essencial determinar els beneficis adequadament i sospesar els riscos abans de procedir amb la prova. Alternativament, algunes parelles trien sotmetre's a tècniques de reproducció assistida amb òvuls donants (recorrent a la fecundació in vitro) o amb esperma donant (inseminació artificial per donant).
Fisiopatologia
La fibrosi quística ocorre quan hi ha una mutació en el gen CFTR. La proteïna creada per aquest gen (proteïna reguladora de la conductància transmembrana de la fibrosi quística o CFTR) s'uneix a la membrana externa de les cèl·lules en les glàndules sudorípares, pulmó, pàncrees, i altres òrgans afectats. La proteïna travessa aquesta membrana i actua com un canal iònic connectant la part interna de la cèl·lula (citoplasma) amb el fluid extracel·lular. Aquest canal és sobretot responsable de controlar el pas de clorur cap a (i des de) el mitjà intern. Quan la proteïna CFTR no funciona correctament, aquest moviment es veu restringit, i es reté clorur en l'espai extracel·lular. Degut al fet que el clorur té càrrega elèctrica negativa, els ions amb càrrega positiva tampoc podran creuar la membrana citoplasmàtica, a causa de l'atracció electroestàtica exercida pels ions clorur. El sodi és el més comú entre els ions presents fora de la cèl·lula, i la combinació de sodi i clorur dona lloc al clorur de sodi, el qual es perd en grans quantitats en la suor dels individus amb FQ. Aquesta pèrdua de sal és l'argument bàsic per explicar l'eficàcia del test de la suor.[4]
El mecanisme pel qual aquesta disfunció cel·lular produeix les manifestacions clíniques abans descrites no es coneix amb exactitud. Una de les teories que intenta explicar-ho suggereix que la falla de la proteïna CFTR per transportar el clorur determina l'acumulació d'abundant moc en els pulmons, creant un mitjà propici (ric en nutrients) per als bacteris, que poden així eludir el sistema immunitari. També es postula que aquesta anomalia en la proteïna CFTR indueix un augment paradoxal en la captura de sodi i clorur, cosa que estimula la reabsorció d'aigua i resulta en la formació de la mucositat deshidratada i espessa. Altres teories s'enfoquen en el fenomen del moviment de clorur cap a l'exterior de la cèl·lula, que també provoca dessecament del moc i de les secrecions pancreàtiques i biliars. En general, aquestes hipòtesis coincideixen a atribuir els trastorns més grans a l'obstrucció dels conductes més prims per les secrecions espesses i glutinoses en els diferents òrgans afectats. Aquesta situació condiciona la infecció crònica i promou la remodelació estructural del pulmó, a més de produir dany pancreàtic (intervingut pels enzims digestius aglomerades), i obstrucció dels intestins per grans bitlles fecals.[4]
Paper de la infecció crònica en la malaltia pulmonar

Micrografia electrònica d'escombrat de bacteri Pseudomonas aeruginosa, associada amb freqüència a les infeccions pulmonars greus que compliquen la FQ.
Els pulmons de les persones amb fibrosi quística són colonitzats i infectats per bacteris des d'edats primerenques. Els microorganismes que es propaguen en aquests pacients prosperen en el moc anòmal acumulat en les vies respiratòries més estretes. El moc glutinós estimula el desenvolupament de microambients bacterians (biofilms) que resulten difícils de penetrar per a les cèl·lules immunes i els antibiòtics. Per la seva banda, els pulmons responen al dany continu, infligit per les secrecions espesses i les infeccions cròniques, remodelant gradualment les vies respiratòries inferiors (bronquiectàsia), cosa que torna la infecció encara més difícil d'erradicar.[25]
Amb el pas del temps canvien tant el tipus de bacteris que afecten a aquests pacients, com les característiques específiques amb què es presenten. En una primera etapa, certs bacteris ordinaris com Staphylococcus aureus i Haemophilus influenzae colonitzen i infecten els pulmons. Més tard, no obstant, prevalen Pseudomonas aeruginosa (i, de vegades, el complex Burkholderia cepacia, integrat per diferents espècies de Burkholderia). Una vegada disseminats per les vies respiratòries, aquests bacteris s'adapten al mitjà i desenvolupen resistència als antibiòtics convencionals. Pseudomonas pot adquirir certes característiques especials, donant lloc a la formació de grans colònies —aquests ceps són coneguts com a Pseudomonas "mucoide" i són rares en persones lliures de la malaltia.[25]
Una de les maneres per la qual la infecció es propaga és per transmissió entre individus amb FQ.[26] En el passat, era habitual que aquests participessin, en forma conjunta, de campaments estiuencs i altres activitats d'esplai.[27][28] Els hospitals allotjaven als pacients amb FQ en una àrea comuna, i l'equipament de rigor (per exemple, els nebulitzadors)[29] no era esterilitzat entre usos successius.[30] Això va conduir a la transmissió de ceps bacterians molt perillosos entre grups de pacients. Actualment, la rutina en establiments d'atenció sanitària consisteix a aïllar a aquests pacients els uns dels altres; a més, el personal a càrrec de la seva cura ha de vestir bates i guants per limitar la proliferació de ceps bacterians virulents.[31] Amb freqüència, els pacients afectats per bacteris particularment perillosos reben atenció en dies i en edificis diferents als assignats a qui no tenen aquestes infeccions.
Biologia molecular
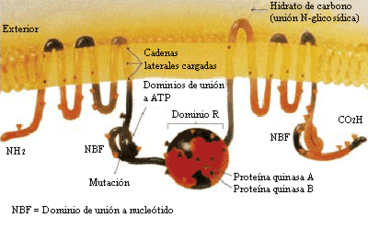
Proteïna CFTR - estructura molecular de la proteïna
La mutació més comuna, ΔF508, és una deleció (Δ) de tres nucleòtids que resulta en una pèrdua de l'aminoàcid fenilalanina (F) en la posició cinc-centena vuitena (508) de la proteïna. Aquesta mutació és responsable del 70% dels casos de FQ a nivell mundial i del 90% en els Estats Units. No obstant això, existeixen més de 1.000 mutacions diferents que poden produir FQ. Són diversos els mecanismes pels quals aquestes mutacions causen problemes en la proteïna CFTR. En particular, la mutació ΔF508 genera una proteïna que no es plega de manera normal i acaba sent degradada per la cèl·lula. Diverses mutacions comunes en la població asquenazita donen lloc a la síntesi de proteïnes massa curtes a causa d'una conclusió anticipada de la seva producció. Altres mutacions menys freqüents originen proteïnes que no utilitzen l'energia com cal, que no permeten que el clorur creui la membrana apropiadament, o que són degradades a una taxa més ràpida que la normal. Certes mutacions poden conduir també a un minvament en la producció de còpies de la proteïna CFTR.[4]
Estructuralment, el gen CFTR pertany a la denominada superfamília de transportadors ABC (acrònim per a l'anglès ATP Binding Cassette, "casset d'unió a l'ATP").[4] L'estructura terciària de la proteïna codificada per aquest gen consta de dos dominis capaços d'hidrolitzar adenosín trifosfat, cosa que permet a la proteïna utilitzar energia en la forma d'ATP. Així mateix, un altre parell de dominis, cadascun constituït per sis hèlixs alfa, possibilita el pas de la proteïna a través de la membrana cel·lular. L'activació es concreta per reacció de fosforilació en un lloc d'unió regulador, sobretot mitjançant la proteïna-cinasa A (PKA, abans denominada cAPK o proteïna cinasa dependent de l'adenosina monofosfat cíclic (AMPc)).[4] El carboxil terminal (C-) de la proteïna està unit al citoesquelet per interacció amb dominis proteics PDZ>.[32]
Tractament
Un aspecte fonamental en la terapèutica de la fibrosi quística és el control i el tractament del dany pulmonar causat pel moc espès i per les infeccions, amb l'objectiu de millorar la qualitat de vida del pacient. Per al tractament de les infeccions cròniques i agudes s'administren antibiòtics per vies intravenosa, inhalatòria i oral. També s'utilitzen dispositius mecànics i fàrmacs (en forma d'inhaladors) per controlar les secrecions, i d'aquesta manera descongestionar i desobstruir les vies respiratòries. Altres aspectes de la teràpia es relacionen amb el tractament de la diabetis amb insulina, de la malaltia pancreàtica amb reemplaçament enzimàtic, i de la infertilitat amb tècniques reproductives avançades. Addicionalment, es postula l'eficàcia de diferents procediments, com el trasplantament i la teràpia gènica, per resoldre alguns dels efectes associats a aquesta malaltia.
Antibiòtics per tractar la malaltia pulmonar
Els antibiòtics es prescriuen sempre que existeixi sospita de pneumònia o es constati deteriorament de la funció pulmonar. Habitualment, se'ls escull en funció de l'historial d'infeccions que van afectar al pacient prèviament. Molts dels bacteris comuns en la fibrosi quística són resistents a gran quantitat d'antibiòtics i requereixen setmanes de tractament intravenós amb vancomicina, tobramicina, meropenem, ciprofloxacina i piperacilina.

PICC o catèter central inserit percutàniament (radiografia de tòrax)
La teràpia perllongada sovint requereix hospitalització i canalització d'una via intravenosa permanent com, per exemple, un catèter central inserit percutàniament (PICC). Així mateix, és freqüent la indicació simultània d'antibiòtics administrats per inhalació, com la tobramicina, la colistina i la gentamicina, per diversos mesos, amb l'objectiu de millorar la funció pulmonar impedint la proliferació bacteriana.[33][34] Alguns antibiòtics orals com la ciprofloxacina o l'azitromicina s'utilitzen de vegades per ajudar a prevenir la infecció o per controlar-la una vegada que ja està en curs.[35] En alguns casos passen anys entre successives hospitalitzacions, mentre que en altres es requereix cada any per poder realitzar el tractament.
En tractaments perllongats, alguns dels antibiòtics més comuns (com la tobramicina i la vancomicina) poden causar pèrdua d'audició per ototoxicitat o problemes en els ronyons. Per prevenir tals efectes secundaris, és habitual amidar quantitativament les concentracions d'aquests medicaments en sang i, si és necessari, ajustar les dosis.
Altres mètodes per tractar la malaltia pulmonar

Inhalador típic de salmeterol, agent simpaticomimètic que dilata la llum dels bronquis.
Són diverses les tècniques que s'implementen amb l'objectiu de fluidificar l'esput i facilitar-ne l'espectoració. En el mitjà hospitalari es fa servir la fisioteràpia: un terapeuta practica una sèrie de maniobres mitjançant pressions i percussions (palmoteig) exercides sobre l'exterior del pit (tòrax) diverses vegades al dia. Els dispositius mecànics que actuen sota el mateix principi que aquelles tècniques bàsiques de drenatge postural inclouen el ventilador d'alta freqüència oscil·latòria i els aparells de ventilació percussiva intrapulmonar, dels quals existeixen models portàtils adaptables a l'ús en la casa del pacient.[36] L'exercici aeròbic és altament beneficiós per a les persones amb fibrosi quística, ja que no només promou la descongestió de l'esput, sinó que millora la salut cardiovascular i l'estat general.
Aparell de ventilació percussiva intrapulmonar.
Entre les substàncies administrades per inhalació que ajuden a alleugerir les secrecions i faciliten la seva expulsió, es troben la dornasa alfa i la solució salina hipertònica.[37] La dornasa és una desoxirribonucleasa (ADNasa o DNasa) humana recombinant, que descompon l'ADN en l'esput, reduint així la viscositat d'aquest últim.[38] L'N-acetilcisteïna (un derivat de l'aminoàcid cisteïna) també actua fluidificant l'esput, però les investigacions i l'experiència disponibles han demostrat que els beneficis són poc significatius. Finalment, broncodilatadors com el salbutamol i el salmeterol (ambdós agents, agonistes β2-adrenèrgics) o el bromur de ipratropi (un antagonista del receptor colinèrgic, derivat quaternari de l'atropina) s'utilitzen per augmentar la grandària de les vies respiratòries més petites, al relaxar el múscul llis bronquial.
En la mesura que s'agreuja la condició pulmonar, pot requerir-se suport respiratori mecànic. Per les nits, alguns pacients han d'usar màscares especials que actuen empenyent el flux aeri fins als pulmons. La ventilació no invasiva mitjançant màscara nasal i pressió positiva (VPAP, per l'acrònim per a l'anglès variable positive airway pressure), ajuda a prevenir, durant el somni, caigudes significatives en els nivells sanguinis d'oxigen. També pot usar-se en el curs de la fisioteràpia respiratòria per afavorir l'expulsió d'esput.[39] No obstant això, en casos severs, pot ser necessari implementar formes invasives d'assistència respiratòria amb intubació endotraqueal (això és, col·locació d'un tub o sonda a la tràquea).
Tractament d'altres aspectes de la FQ

La injecció intracitoplasmàtica d'esperma (ICSI, per les seves sigles en anglès) sol indicar-se per a revertir la infertilitat en homes amb FQ.
Els nounats amb ili meconial típicament requereixen cirurgia; en general, no succeeix el mateix en adults amb síndrome d'obstrucció intestinal distal. El tractament de la insuficiència pancreàtica basat en reemplaçament dels enzims digestius minvats permet que els intestins absorbeixin de manera apropiada nutrients i vitamines que, d'una altra manera, es perdrien en la femta. Tanmateix, la majoria dels individus amb FQ han de rebre dosis addicionals de vitamines A, D, I i K a partir de suplements, i seguir una dieta d'alt valor calòric.
La diabetis que sol acompanyar la FQ es tracta amb injeccions d'insulina.[40] El desenvolupament d'osteoporosi pot prevenir-se amb suplements de vitamina D i calci, i sovint es tracta amb bifosfonats.[41]
Quant al retard en el creixement, es procura contrarestar mitjançant la inserció d'un tub d'alimentació (gastrostomia) per augmentar així la ingesta de calories a partir de nutrició addicional; també s'administren a aquest efecte injeccions d'hormona de creixement.[42]
Les infeccions dels sins paranasals solen tractar-se amb un perllongat règim d'antibiòtics. El desenvolupament de pòlips, així com altres canvis estructurals de tipus patològic en l'interior dels conductes nasals, poden restringir el flux aeri i complicar el quadre. Per aquest motiu, és freqüent la pràctica quirúrgica que procura alleujar l'obstrucció i limitar el desenvolupament de noves infeccions. També s'administren corticosteroides intranasals, com la fluticasona, per reduir la inflamació.[43]
Pel que fa a la infertilitat femenina, es pot combatre recorrent a tècniques de reproducció assistida. La infertilitat masculina també té tractament, per exemple, mitjançant la injecció intracitoplasmàtica d'esperma.[44]
Trasplantament i teràpia gènica
En general, es considera procedent el trasplantament de pulmó en persones amb deterioració progressiva de la funció pulmonar i creixent intolerància a l'exercici (fatiga o esgotament muscular desproporcionats per a l'exercici realitzat). Encara que el trasplantament d'un únic pulmó és viable en altres malalties, en els pacients amb FQ ambdós han de ser reemplaçats, ja que, d'una altra manera, els bacteris allotjats en l'òrgan romanent podrien infectar el que ha estat trasplantat. Així mateix, pot practicar-se simultàniament un trasplantament de pàncrees o de fetge amb el propòsit d'alleujar la malaltia hepàtica o la diabetis.[45] L'opció del trasplantament de pulmó s'avalua quan la funció pulmonar es veu afectada en grau tal que es vegi amenaçada la supervivència o es requereixi l'assistència amb dispositius mecànics.[46]
Els adenovirus poden ser modificats per a utilitzar-se en teràpia gènica.
La teràpia gènica representa una via promissòria en la lluita contra la malaltia. Mitjançant aquesta tècnica es procura inserir una còpia normal del gen CFTR en les cèl·lules afectades. A causa de la incapacitat dels retrovirus per arribar a cèl·lules que no es divideixen, s'han realitzat anàlisis clíniques per a inserir gens en adenovirus. En l'actualitat, aquests virus s'estan utilitzant en assajos en els quals el gen CFTR normal s'administra, per un mètode en aerosol, a les cèl·lules epitelials que revesteixen els pulmons (teràpia gènica in vivo). S'espera que els adenovirus insereixin el gen normal, induint una funció pertinent dels canals de clor en aquestes cèl·lules.
Alguns estudis han assenyalat que per prevenir les manifestacions pulmonars de la fibrosi quística, només es requereix l'expressió gènica d'entre un 5 i un 10% dels valors normals de proteïna CFTR.[47] Un inconvenient dels adenovirus és que no s'integren en l'ADN de la cèl·lula hoste. Per tant, finalment es perden, originant una expressió del gen transitòria i la necessitat de reintroducció del vector. S'han proposat diversos abordatges i s'han iniciat nombrosos estudis clínics però, a data de 2006, persisteixen múltiples obstacles que caldrà superar perquè la teràpia gènica resulti reeixida.[48]
Epidemiologia

Herència mendeliana autosòmica recessiva: dues mutacions de línia germinal (una de cadascun dels pares) per a desenvolupar la malaltia; igualment transmesa per homes i dones.
La fibrosi quística és, entre les persones d'ascendència europea, la més freqüent de les malalties autosòmiques recessives potencialment fatals. En els Estats Units, aproximadament 30.000 individus pateixen FQ; en la seva majoria són diagnosticats als sis mesos d'edat. Canadà té prop de 3.000 habitants amb aquesta condició. S'estima que una de cada 25 persones d'ascendència europea i una de cada 29 persones d'ascendència asquenazita són portadors d'una mutació de fibrosi quística. Encara que és menys comuna en aquests grups, aproximadament un de cada 46 hispanoamericans, un de cada 65 africans i un de cada 90 asiàtics són portadors d'almenys un gen CFTR anormal.[49][50][51]Argentina representa una excepció en el context d'Amèrica Llatina, amb una incidència de casos molt major a la mitjana de la regió i molt pròxima a la registrada en els Estats Units o Canadà, i una prevalença de portadors sans en la població general d'1 en 30.
La fibrosi quística es diagnostica tant en homes com en dones. Per raons que només en part es coneixen, l'esperança de vida al néixer resulta ser major entre els barons afectats que entre les dones.[52] Aquest indicador tendeix a variar principalment en funció de l'abast i la qualitat de l'atenció subministrada pels sistemes de salut pública. El 1959, la supervivència mitjana en nens amb FQ era de 6 mesos. Per als nascuts el 2006 als Estats Units, aquest valor grimparia als 36,8 anys, d'acord a les dades compilades per la Fundació de la Fibrosi Quística.[2] La taxa d'esperança de vida ha evolucionat de forma anàloga per a bona part d'Occident, exceptuant els països menys desenvolupats, on es reporten xifres sensiblement menors, i en els quals la majoria de la població afectada no sobreviu més enllà dels deu anys.
La Fundació de la Fibrosi Quística compila, a més, informació sobre l'estil de vida dels adults nord-americans amb FQ. El 2004, la fundació va reportar que el 91% d'aquesta població havia completat l'ensenyament mitjà, i el 54% havia accedit a alguna forma d'educació universitària. Les dades en matèria d'ocupació van revelar que el 12,6% d'aquests adults estava impossibilitat per treballar (quedant fora de la població econòmicament activa), i el 9,9% estava desocupat. D'altra banda, la informació marital va assenyalar que un 59% era solter i un 36% estava casat o vivint en parella. El 2004, 191 dones amb FQ es trobaven embarassades als Estats Units.[53]
Teories sobre la prevalença de la FQ
S'estima que la mutació ΔF508 pot tenir fins a uns 52.000 anys d'antiguitat.[54] S'han formulat nombroses hipòtesis intentant explicar perquè una mutació letal com aquesta ha persistit i s'ha estès entre la població humana. Algunes malalties autosòmiques recessives comunes com l'anèmia falciforme han revelat la propietat de protegir als seus portadors d'altres afeccions, concepte conegut com a avantatge heterozigota. Amb el descobriment que la toxina del còlera requereix que les seves hostes siguin proteïnes CFTR normals per a poder funcionar apropiadament, s'ha postulat que els portadors de gens CFTR mutants van obtenir el benefici de la resistència al còlera i a altres causes de diarrea.[55] No obstant això, estudis posteriors no han confirmat aquesta hipòtesi.[56][57]
La presència de proteïnes CFTR normals és condició necessària per a l'ingrés de Salmonella typhi (serotip de Salmonella enterica, proteobacteri gram negatiu del gènere Salmonella) en les cèl·lules,[58] el que suggereix que els portadors de gens CFTR mutants podrien ser resistents a la febre tifoide. No obstant això, cap estudi in vivo ha confirmat aquesta hipòtesi. En qualsevol dels casos, la baixa incidència de fibrosi quística fora d'Europa, en llocs on tant el còlera com la febre tifoide són endèmics, manca d'explicació immediata.
Història
Encara que l'espectre clínic complet de la FQ no va ser reconegut fins als anys 1930, certs aspectes van ser identificats molt abans. Carl von Rokitansky va descriure un cas de mort fetal amb peritonitis mecònica, una complicació de l'ili mecònic associat amb la fibrosi quística. L'ili mecònic va ser descrit per primera vegada el 1905 per Karl Landsteiner.[59]

Dorothy H. Andersen va descriure per primer cop la fibrosi quística (National Library of Medicine).
El 1938, Dorothy Andersen va publicar un article intitulat Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathological study
("La fibrosi quística del pàncrees i la seva relació amb la malaltia celíaca: un estudi clínic i patològic") a la revista American Journal of Diseases of Children. D'aquesta manera, era la primera investigadora a definir aquesta entitat nosològica (denominada, per aquell temps, "fibrosi quística del pàncrees"), i en correlacionar-la amb els trastorns pulmonars i intestinals prominents.[3] També va postular que era una malaltia recessiva i va utilitzar el reemplaçament d'enzims pancreàtics com tractament per als nens afectats. El 1952, Paul di Sant' Agnese va descobrir anomalies en els electròlits de la suor. Sobre la base d'aquesta evidència, es va desenvolupar i va perfeccionar l'examen de la suor durant el curs de la següent dècada.[60]
El 1985, investigadors de Londres, Toronto i Salt Lake City van traçar el mapa del gen CFTR en el cromosoma 7q. Quatre anys més tard, el 1989, Francis Collins, Lap-Chee Tsui i John R. Riordan van descobrir la primera mutació per a la FQ, ΔF508, en aquest cromosoma. Investigacions posteriors a aquella troballa, van identificar més de mil mutacions diferents que donen origen a la malaltia. Lap-Chee Tsui va liderar l'equip de científics de l'Hospital for Sick Children (un hospital escola en conveni amb la Universitat de Toronto) que va descobrir el gen responsable de la FQ. Es tracta del primer trastorn genètic dilucidat estrictament mitjançant el procés de genètica inversa. Degut al fet que les mutacions del gen CFTR són generalment petites, les tècniques de la genètica clàssica o formal no van ser capaces de determinar amb precisió el gen mutant.[61] Utilitzant marcadors proteics, els estudis de lligamient genètic van aconseguir traçar un mapa de la mutació del cromosoma 7. Les tècniques de passeig i salt cromosòmics van servir llavors per identificar i seqüenciar el gen.[62]
La identificació de la mutació específica responsable de la FQ en un pacient pot ser útil per predir l'evolució de la malaltia. Per exemple, els pacients homozigots per a la mutació ΔF508 presenten, en gairebé tots els casos, insuficiència pancreàtica i tenen, en general, un grau relativament sever d'afectació respiratòria. No obstant això, existeixen excepcions que indiquen la possibilitat que factors addicionals (potser, gens en altres loci) intervinguin en l'expressió de la malaltia. D'altra banda, la clonació del gen de la FQ ha obert la possibilitat de la teràpia gènica, tal com s'ha descrit en la secció pertinent.
En l'actualitat, l'esperança de vida dels malalts és de 32 anys, tot i que la mitjana de supervivència amb la mutació més comuna, del parell delF508/delF508 és de 23 anys.[63]
Notes
↑ Jorde, Lynn; Carey, John; White, Raymond. Genética médica. Madrid: Mosby, 1996. ISBN 84-8174-161-2
↑ 2,02,1 New Statistics Show CF Patients Living Longer Cystic Fibrosis Foundation (26 d'abril, 2006). Consultat el 26-07-2006. Error de citació: Etiqueta<ref>no vàlida; el nom «median» està definit diverses vegades amb contingut diferent.
↑ 3,03,1 Andersen DH. "Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathological study." Am J Dis Child 1938; 56:344-399
↑ 4,04,14,24,34,44,5 Rowe SM, Miller S, Sorscher EJ. "Cystic fibrosis." N Engl J Med. 2005 may 12;352(19):1992-2001. PMID: 15888700
↑ Maldonado M, Martínez A, Alobid I, Mullol J. The antrochoanal polyp. Rhinology. 2004 dic;42(4):178-82. Rev. PMID: 15626248
↑ Ramsey B, Richardson MA. Impact of sinusitis in cystic fibrosis. Allergy Clin Immunol. 1992 sep;90(3 Pt 2):547-52. PMID: 1527348
↑ Eggermont E, De Boeck K. Small-intestinal abnormalities in cystic fibrosis patients. Eur J Pediatr. 1991 oct;150(12):824-8. Rev. PMID: 1743211
↑ Kulczycki LL, Shwachman H. "Studies in cystic fibrosis of the pancreas; occurrence of rectal prolapse." N Engl J Med. 1958 ag 28;259(9):409-12. PMID: 13578072
↑ Cohn JA, Friedman KJ, Noone PG, Knowles MR, Silverman LM, Jowell PS. Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. N Engl J Med. 1998 sep 3;339(10):653-8. PMID: 9725922
↑ Malfroot A, Dab I. New insights on gastro-oesophageal reflux in cystic fibrosis by longitudinal follow up. Arch Dis Child. 1991 nov;66(11):1339-45. PMID: 175564
↑ Khoshoo V, Udall JN Jr. Meconium ileus equivalent in children and adults. Am J Gastroenterol. 1994 feb;89(2):153-7. PMID: 8304294
↑ Williams SG, Westaby D, Tanner MS, Mowat AP. Liver and biliary problems in cystic fibrosis. Br Med Bull. 1992 oct;48(4):877-92. PMID: 1458306
↑ Moran A, Pyzdrowski KL, Weinreb J, Kahn BB, Smith SA, Adams KS, Seaquist ER. "Insulin sensitivity in cystic fibrosis." Diabetes. Agosto 1994;43(8):1020-6. PMID: 8039595
↑ Haworth CS, Selby PL, Webb AK, Dodd ME, Musson H, McL Niven R, Economou G, Horrocks AW, Freemont AJ, Mawer EB, Adams JE. "Low bone mineral density in adults with cystic fibrosis." Thorax. 1999 nov;54(11):961-7. PMID: 10525552
↑ McCallum TJ, Milunsky JM, Cunningham DL, Harris DH, Maher TA, Oates RD. "Fertility in men with cystic fibrosis: an update on current surgical practices and outcomes." Chest. 2000 oct;118(4):1059-62. PMID: 11035677
↑ Dodge JA. "Male fertility in cystic fibrosis." Lancet. 1995 sep 2;346(8975):587-8. PMID: 7650999
↑ Augarten A, Yahav Y, Kerem B, Halle D, Laufer J, Szeinberg A, Dor J, Mashiach S, Gazit E, Madgar I. "Congenital bilateral absence of vas deferens in the absence of cystic fibrosis." Lancet 344: 1473-74, 1994. PMID: 7968122
↑ Gilljam M, Antoniou M, Shin J, Dupuis A, Corey M, Tullis DE. "Pregnancy in cystic fibrosis. Fetal and maternal outcome." Chest. 2000 jul;118(1):85-91. PMID: 10893364
↑ Gibson LE, Cooke RE. "A test for concentration of electrolytes in sweat in cystic fibrosis of the pancreas utilising pilocarpine by iontophoresis." Pediatrics mar;23(3):545-9. PMID: 13633369
↑ Stern, RC. "The diagnosis of cystic fibrosis." N Engl J Med 1997; 336:487. PMID: 9017943
↑ ACOG Committee Opinion #325: "Update on Carrier Screening for Cystic Fibrosis". Obstet Gynecol 2005; 106:1465.
↑ American College of Obstetricians and Gynecologists and American College of Medical Genetics. Preconception and prenatal carrier screening for cystic fibrosis. Clinical and laboratory guidelines. American College of Obstetricians and Gynecologists, Washington, DC, octubre 2001.
↑ Elias, S, Annas, GJ, Simpson, JL. Carrier screening for cystic fibrosis: Implications for obstetric and gynecologic practice. Am J Obstet Gynecol 1991; 164:1077. PMID: 2014829
↑ Tabor A, Philip J, Madsen M, Bang J, Obel EB, Norgaard-Pedersen B. Randomised controlled trial of genetic amniocentesis in 4606 low-risk women. Lancet. 1986 jun 7;1(8493):1287-93. PMID: 2423826
↑ 25,025,1 Saiman L. Microbiology of early CF lung disease. Paediatr Respir Rev. 2004;5 Supl A:S367-9. PMID: 14980298
↑ Tummler B, Koopmann U, Grothues D, Weissbrodt H, Steinkamp G, von der Hardt H. Nosocomial acquisition of Pseudomonas aeruginosa by cystic fibrosis patients. J Clin Microbiol. 1991 jun;29(6):1265-7. PMID: 1907611
↑ Centers for Disease Control and Prevention (CDC). Pseudomonas cepacia at summer camps for persons with cystic fibrosis. MMWR Morb Mortal Wkly Rep. 1993 jun 18;42(23):456-9. PMID: 7684813
↑ Pegues DA, Carson LA, Tablan OC, FitzSimmons SC, Roman SB, Miller JM, Jarvis WR.Acquisition of Pseudomonas cepacia at summer camps for patients with cystic fibrosis. Summer Camp Study Group. J Pediatr. 1994 may;124(5 Pt 1):694-702. PMID: 7513755
↑ Pankhurst CL, Philpott-Howard J. The environmental risk factors associated with medical and dental equipment in the transmission of Burkholderia (Pseudomonas) cepacia in cystic fibrosis patients. J Hosp Infect. 1996 Apr;32(4):249-55. PMID: 8744509
↑ Jones AM, Govan JR, Doherty CJ, Dodd ME, Isalska BJ, Stanbridge TN, Webb AK. Identification of airborne dissemination of epidemic multiresistant strains of Pseudomonas aeruginosa at a CF centre during a cross infection outbreak. Thorax. 2003 jun;58(6):525-7 PMID: 12775867
↑ Hoiby N. Isolation and treatment of cystic fibrosis patients with lung infections caused by Pseudomonas (Burkholderia) cepacia and multiresistant Pseudomonas aeruginosa. Neth J Med. 1995 jun;46(6):280-7. PMID: 7643943
↑ Short DB, Trotter KW, Reczek D, Kreda SM, Bretscher A, Boucher RC, Stutts MJ, Milgram SL. An apical PDZ protein anchors the cystic fibrosis transmembrane conductance regulator to the cytoskeleton. J Biol Chem. 1998 jul 31;273(31):19797-801. PMID: 9677412
↑ Pai VB, Nahata MC. Efficacy and safety of aerosolized tobramycin in cystic fibrosis. Pediatr Pulmonol. 2001 oct;32(4):314-27. Rev. PMID: 11568993
↑ Westerman EM, Le Brun PP, Touw DJ, Frijlink HW, Heijerman HG. Effect of nebulized colistin sulphate and colistin sulphomethate on lung function in patients with cystic fibrosis: a pilot study. J Cyst Fibros. 2004 mar;3(1):23-8. PMID: 15463883
↑ Hansen CR, Pressler T, Koch C, Hoiby N.Long-term azithromycin treatment of cystic fibrosis patients with chronic Pseudomonas aeruginosa infection; an observational cohort study. J Cyst Fibros. 2005 mar;4(1):35-40. PMID: 15752679
↑ van der Schans C, Prasad A, Main E. Chest physiotherapy compared to no chest physiotherapy for cystic fibrosis. Cochrane Database Syst Rev. 2000;(2):CD001401. Rev. PMID: 10796781
↑ Kuver R, Lee SP. Hypertonic saline for cystic fibrosis. N Engl J Med. 2006 abr 27;354(17):1848-51; resposta de l'autor 1848-51. PMID: 16642591
↑ Lieberman J. "Dornase aerosol effect on sputum viscosity in cases of cystic fibrosis." JAMA. 1968 jul 29;205(5):312-3. PMID: 5694947
↑ Moran F, Bradley J. Non-invasive ventilation for cystic fibrosis. Cochrane Database Syst Rev. 2003;(2):CD002769. Rev. PMID: 12804435
↑ Onady GM, Stolfi A. Insulin and oral agents for managing cystic fibrosis-related diabetes. Cochrane Database Syst Rev. 2005 jul 20;(3):CD004730. Rev. PMID: 16034943
↑ Conway SP, Oldroyd B, Morton A, Truscott JG, Peckham DG. Effect of oral bisphosphonates on bone mineral density and body composition in adult patients with cystic fibrosis: a pilot study. Thorax. 2004 ag;59(8):699-703. PMID: 15282392
↑ Hardin DS, Rice J, Ahn C, Ferkol T, Howenstine M, Spears S, Prestidge C, Seilheimer DK, Shepherd R. Growth hormone treatment enhances nutrition and growth in children with cystic fibrosis receiving enteral nutrition.J Pediatr. 2005 mar;146(3):324-8 PMID: 15756212
↑ Marks SC, Kissner DG. Management of sinusitis in adult cystic fibrosis. Am J Rhinol. 1997 en-feb;11(1):11-4. PMID: 9065342
↑ Phillipson GT, Petrucco OM, Matthews CD. Congenital bilateral absence of the vas deferens, cystic fibrosis mutation analysis and intracytoplasmic sperm injection. Hum Reprod. 2000 feb;15(2):431-5. PMID: 10655317
↑ Simultaneous liver and pancreas transplantation in patients with cystic fibrosis. Transplant Proc. 2005 oct;37(8):3567-9. PMID: 16298663
↑ Belkin RA, Henig NR, Singer LG, Chaparro C, Rubenstein RC, Xie SX, Yee JY, Kotloff RM, Lipson DA, Bunin GR. Risk factors for death of patients with cystic fibrosis awaiting lung transplantation. Am J Respir Crit Care Med. 2006 mar 15;173(6):659-66. Epub 2005 dic 30. PMID: 16387803
↑ Ramalho AS, Beck S, Meyer M, Penque D, Cutting GR, Amaral MD. Five percent of normal cystic fibrosis transmembrane conductance regulator mRNA ameliorates the severity of pulmonary disease in cystic fibrosis. Am J Respir Cell Mol Biol. 2002 nov;27(5):619-27. PMID: 12397022
↑ Tate S, Elborn S. Progress towards gene therapy for cystic fibrosis.Expert Opin Drug Deliv. 2005 mar;2(2):269-80. Rev. PMID: 16296753
↑ Rosenstein BJ and Cutting GR. The diagnosis of cystic fibrosis: a consensus statement. Cystic Fibrosis Foundation Consensus Panel. J Pediatr. 1998 abr;132(4):589-95. Rev. PMID: 9580754
↑ Hamosh A, Fitz-Simmons SC, Macek M Jr, Knowles MR, Rosenstein BJ, Cutting GR. Comparison of the clinical manifestations of cystic fibrosis in black and white patients. J Pediatr. 1998 feb;132(2):255-9. PMID: 9506637
↑ Kerem B, Chiba-Falek O, Kerem E. Cystic fibrosis in Jews: frequency and mutation distribution. Genet Test. 1997;1(1):35-9. Rev. PMID: 10464623
↑ Rosenfeld, M, Davis, R, FitzSimmons, S, i cols. Gender gap in cystic fibrosis mortality. Am J Epidemiol 1997 145,794-803
↑ Cystic Fibrosis Foundation PDF
PDF
↑ Wiuf C. Do delta F508 heterozygotes have a selective advantage? Genet Res. 2001 ag;78(1):41-7. PMID: 11556136
↑ Gabriel SE, Brigman KN, Koller BH, Boucher RC, Stutts MJ. Cystic fibrosis heterozygote resistance to cholera toxin in the cystic fibrosis mouse model. Science. 1994 oct 7;266(5182):107-9. PMID: 7524148
↑ Cuthbert AW, Halstead J, Ratcliff R, Colledge WH, Evans MJ. The genetic advantage hypothesis in cystic fibrosis heterozygotes: a murine study. J Physiol. 1995 enero 15;482 (Pt 2):449-54. PMID: 7714835
↑ Hogenauer C, Santa Ana CA, Porter JL, Millard M, Gelfand A, Rosenblatt RL, Prestidge CB, Fordtran JS. Active intestinal chloride secretion in human carriers of cystic fibrosis mutations: an evaluation of the hypothesis that heterozygotes have subnormal active intestinal chloride secretion. Am J Hum Genet. 2000 dic;67(6):1422-7. Epub 2000 oct 30. PMID: 11055897
↑ Pier GB, Grout M, Zaidi T, Meluleni G, Mueschenborn SS, Banting G, Ratcliff R, Evans MJ, Colledge WH. Salmonella typhi uses CFTR to enter intestinal epithelial cells. Nature. 1998 may 7;393(6680):79-82. PMID: 9590693
↑ Busch R. "On the history of cystic fibrosis." Acta Univ Carol [Med] (Praga). 1990;36(1-4):13-5. PMID: 2130674
↑ Di Sant' Agnese PA, Darling RC, Perera GA, i cols. Abnormal electrolyte composition of sweat in cystic fibrosis of the pancreas: clinical implications and relationship to the disease. Pediatrics 1953; 12:549-563.
↑ Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, i cols. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989 sep 8;245(4922):1066-73. Erratum in: Science 1989 sep 29;245(4925):1437. PMID: 2475911
↑ Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N, i cols. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989 sep 8;245(4922):1059-65. PMID: 2772657
↑ Harrison, Margaret A. Progress in cystic fibrosis research (en anglès). Nova Publishers, 2005, p.1. ISBN 1594542325.
Bibliografia
| A Wikimedia Commons hi ha contingut multimèdia relatiu a: Fibrosi quística |
- Dapena Fernández, Francisco Javier. Fibrosis quística. Salobreña: Alhulia, 1998, 1a ed. ISBN 84-95136-13-9
- Salcedo Posadas, Antonio; García Novo, María Dolores. Fibrosis quística. Madrid: Díaz de Santos, 1998, 1a ed. ISBN 84-7978-368-0
- Segal, Edgardo. Fibrosis quística. Buenos Aires: Journal, 2004, 1a ed. ISBN 987-97739-7-7
- Segal E, i cols. "Consenso de Fibrosis Quística." Arch.argent.pediatr. 1999;97(3):188 PDF


